Genómica: conocimiento
Un enfoque de aprendizaje profundo en torno a la COVID-19 y otras enfermedades infecciosas
Predicción de cambios estructurales en proteínas virales causados por mutaciones genéticas
En la actualidad hay casi 3 millones de casos confirmados de COVID-19 en todo el mundo, lo que provoca cientos de miles de muertes y dificultades económicas. Recientemente, en un estudio que aún no ha sido revisado por colegas, los investigadores de la Universidad de Zhejiang de China afirmaron que el coronavirus había mutado en al menos 30 cepas y que la patogenicidad difería entre cada cepa1. El estudio se presentó como una posible explicación de los distintos niveles de gravedad de los brotes en todo el mundo. A medida que las compañías de biotecnología y los gobiernos se apresuran a desarrollar una vacuna y tratamientos para esta enfermedad altamente contagiosa, este hallazgo plantea la preocupación de que ciertos tratamientos para una cepa pueden no ser efectivos para otras debido a las diferencias en la estructura de las proteínas, como sus receptores. Se han publicado secuencias genómicas completas o parciales para la mayoría de las cepas conocidas de la COVID-19, pero la brecha en el conocimiento científico actualmente radica en cómo estas mutaciones genéticas se traducen en estructuras proteicas físicas. Desarrollar vacunas y tratamientos será mucho más difícil sin una comprensión concreta de las proteínas asociadas.
Una solución a este problema se puede encontrar en el aprendizaje automatizado. El aprendizaje automatizado es una rama de la inteligencia artificial que puede discernir patrones en los datos y usarlos para hacer predicciones sobre nuevos conjuntos de datos. Los investigadores de DeepMind, una compañía de investigación de inteligencia artificial con sede en el Reino Unido, publicaron recientemente un artículo en Nature en el que detallaron un nuevo programa de aprendizaje automatizado llamado AlphaFold2. Este sistema se basa en las secuencias de aminoácidos de las proteínas, las cuales se derivan de las secuencias de ADN conocidas.
Información contextual
Las proteínas son moléculas complejas que son esenciales para todos los organismos biológicos. El ADN funciona como un modelo para construir estas estructuras, donde una transcripción de ARN de tres nucleótidos de ADN codifica un aminoácido específico. Una cadena de aminoácidos se pliega para formar una proteína. La forma en que se pliegan las cadenas de aminoácidos es extremadamente compleja y difícil de predecir. Actualmente, los científicos solo pueden llegar a derivar secuencias de aminoácidos a partir de una línea de código genético. No hay otra forma de observar la intrincada estructura de la proteína que no sea a través de una experimentación que requiere mucho tiempo. Por ejemplo, el 90 % de las estructuras proteicas disponibles en el Banco de Datos de Proteínas se encontraron mediante cristalografía de rayos X, que utiliza la densidad de electrones en tres dimensiones para inferir las posiciones de los átomos3. La principal limitación que impide a los científicos predecir con exactitud todas las estructuras de las proteínas es la falta de un modelo generalmente aceptado para explicar la naturaleza compleja del plegamiento de proteínas.
En el caso de la COVID-19, el problema del plegamiento de proteínas entra en juego de una manera muy impactante. A medida que la COVID-19 muta en diferentes cepas, los cambios en el código genético alteran la secuencia de aminoácidos, que a su vez, influyen en la estructura final de la proteína. Los tratamientos para la COVID-19 se basan en la comprensión completa, por ejemplo, de la estructura de los receptores en la superficie del virus, ya que un fármaco debería poder adherirse a él en un mecanismo de llave y candado y desmovilizarlo. Como tal, es necesario que haya un proceso eficiente mediante el cual se puedan predecir rápidamente estructuras proteicas complejas con base en la publicación de las secuencias genéticas de nuevas cepas de coronavirus.
"A medida que la COVID-19 muta en diferentes cepas, los cambios en el código genético alteran la secuencia de aminoácidos, que a su vez, influyen en la estructura final de la proteína".
Descomposición del sistema AlphaFold
En su artículo, los investigadores de DeepMind explican que AlphaFold se basa en redes neuronales profundas (Deep Neural Network, DNN). Las DNN toman como entrada una secuencia de aminoácidos para una proteína. Luego, generan dos predicciones: la distancia entre los aminoácidos y el ángulo de los enlaces químicos entre ellos. Estos resultados se pueden utilizar para producir un diagrama 3D de la proteína.
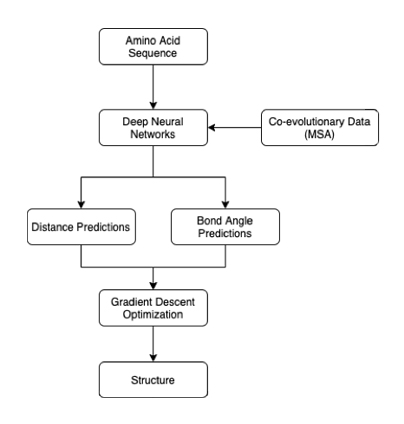
Figura 1: Funcionamiento interno de AlphaFold
AlphaFold se compone de tres redes neuronales: dos encuentran atributos dimensionales de residuos y la tercera combina estas predicciones para resolver qué tan cerca están de la respuesta correcta4. El sistema depende del análisis coevolutivo. El alineamiento de secuencia múltiple (multiple sequence alignment, MSA) destaca las similitudes en las secuencias de aminoácidos en proteínas homólogas. Las similitudes sugieren que estos fragmentos coevolucionaron con el tiempo, lo que luego les permite a los investigadores inferir su proximidad en el espacio. Es importante señalar que este enfoque no debe confundirse con el modelado basado en plantillas, ya que los investigadores simplemente utilizaron datos relacionados con similitudes entre secuencias en vez de la creación a partir de un modelo 3D determinado experimental de otra proteína (la plantilla).
Sin embargo, las distancias y los ángulos de enlace predichos no son necesariamente posibles en una proteína real. Para resolver este problema, los investigadores inicializan una estructura de proteína que es empíricamente posible. Luego, mediante un proceso conocido como descenso de gradiente, la estructura se refina iterativamente para que coincida con la distancia predicha y los ángulos de enlace lo más cerca posible. De esta forma, las estructuras generadas pueden satisfacer los parámetros predichos, sin dejar de ser físicamente posible. El modelo se entrenó en un conjunto de datos de 29,427 proteínas del Banco de Datos de Proteínas.
En términos de rendimiento, AlphaFold superó a sus equivalentes. El Experimento Amplio Comunitario sobre la Evaluación Crítica de Técnicas para la Predicción de la Estructura de las Proteínas (Critical Assessment of Techniques for Protein Structure Prediction, CASP) es una competencia bienal en la que los investigadores concursan para predecir informáticamente la mayor cantidad de proteínas objetivo con la mayor precisión posible. La CASP consta de dos categorías: modelado libre (free modeling, FM) y modelado basado en plantillas (template-based modeling, TBM). La categoría de modelado libre requiere entradas para predecir la estructura de una proteína basándose únicamente en su secuencia, mientras que el modelado basado en plantillas es el proceso de predecir la estructura de una proteína desconocida con base en proteínas que ya se encuentran en el Banco de Datos de Proteínas.
Dos métricas que utiliza la CASP para evaluar la nueva técnica son la puntuación del modelado de plantillas (template modeling, TM) y la puntuación total de la prueba de distancia global (global distance test total score, GDT_TS). Ambas puntuaciones calculan la similitud entre dos estructuras de proteínas. La puntuación del TM mide qué tan cerca un modelo predicho coincide con la estructura real en una escala de 0 a 1. AlphaFold pudo predecir 23 de 43 dominios de modelado libre con una puntuación de TM superior a 0.7, mientras que la siguiente mejor entrada logró solo 14 de 43. Más impresionante aún, AlphaFold aumentó significativamente la tasa de progreso de las competencias anteriores de la CASP. La puntuación total de la prueba de distancia global (GDT_TS) mide la topología bruta de una predicción en comparación con la estructura real en una escala de 0 a 100. AlphaFold prácticamente duplicó el aumento esperado en GDT_TS de CASP12 a CASP135. Las entradas clasificadas en CASP13 se basan en la puntuación z sumada y limitada, que básicamente muestra qué tan lejos del modelo de rendimiento promedio estaba un modelo: las entradas superiores tienen una puntuación z sumada positiva y las entradas inferiores reciben una puntuación z negativa. AlphaFold alcanzó una puntuación z sumada de 52.8 en comparación con la siguiente mejor puntuación 36.6 en la categoría FM. En la categoría TBM, AlphaFold pudo funcionar a la par con otros modelos, a pesar de no usar ninguna plantilla.

Estos avances en el modelado informático del plegamiento de proteínas son muy interesantes. Aunque las predicciones de AlphaFold todavía no son completamente precisas, no hay duda de que tendrán un impacto considerable en el panorama biomédico. Actualmente, otros investigadores han ampliado los métodos presentados por DeepMind para crear modelos aún más precisos. Los investigadores del laboratorio Baker Lab publicaron sus hallazgos sobre una versión mejorada de AlphaFold llamada TrRosetta, que logró una puntuación de TM promedio de 0.625 en comparación con 0.5876 de AlphaFold.6.
En futuros brotes de una enfermedad altamente contagiosa, los programas de predicción de la estructura de proteínas basados en inteligencia artificial (IA) ayudarán a acelerar el descubrimiento de vacunas y tratamientos, además de reducir costos. La determinación experimental de la estructura de la proteína sigue siendo costosa y requiere mucho tiempo. Por ejemplo, la cristalografía de rayos X requiere un equipo costoso y lleva varios meses producir resultados. Por el contrario, el modelado de proteínas basado en IA producirá resultados casi instantáneos y solo requerirá secuencias de ADN y hardware informático suficiente, cuyo precio está bajando rápidamente. Con herramientas de IA como AlphaFold, el tiempo y el costo desde la secuenciación del ADN hasta el desarrollo de vacunas y fármacos disminuirán significativamente.
"El costo de secuenciar un genoma continúa disminuyendo, mientras que el costo de determinar experimentalmente la estructura de la proteína sigue siendo alto y requiere mucho tiempo".
Acerca del autor
Simon Lee es un estudiante de secundaria en Whittle School and Studios en Washington D. C. Actualmente, centra su interés en soluciones basadas en el aprendizaje automatizado para problemas de salud global.