
Regulatory variation is DNA sequence variation that occurs in non-coding genomic regions that influence gene expression. Approximately 93% of disease and trait-associated genome-wide association study (GWAS) variants are located in non-coding regions of the genome, with approximately 20% located 100kb to 1Mb away from any coding sequence1. Therefore, it is important to investigate how non-coding variants influence diseases and traits by linking them to regulatory regions, determining how they are influencing gene expression over long distances and what their target genes are.
"...the genome is tightly packed into the nucleus and takes on a three-dimensional structure in space..."
While we tend to think of the genome as a linear sequence of As, Ts, Cs, and Gs, it is actually tightly packed into the nucleus and takes on a three-dimensional structure in space. Non-coding regulatory elements such as enhancers can regulate the expression level of a gene hundreds of kilobases away through chromosomal looping that can bring distal regulatory elements in three-dimensional proximity to target genes (Figure 1). Variation can occur in the regulatory element that can affect its ability to regulate gene expression, oftentimes by affecting the binding of a transcription factor (Figure 1). For example, one study established evidence that a regulatory region containing prostate cancer-associated variant rs378854 loops over and interacts with the PVT1 oncogene over a distance of 500,000 base pairs2. In the risk allele of this variant, the binding of a repressive transcription factor known as YY1 is reduced and therefore PVT1 expression is increased. On a broader scale, non-coding variants tend to co-localize in regulatory elements. In one study on non-coding variants in inflammatory bowel disease (IBD), 92 of 163 of the non-coding variants are in regulatory elements3. By analyzing these regulatory elements in the context of the 3D genome, the same group of researchers were able to connect these elements to downstream genes. Some of these genes are known to be involved in IBD and some could potentially be novel candidate genes4.
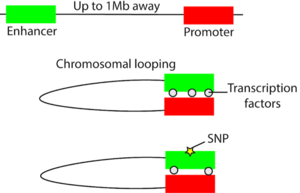
Figure 1: Disruption of chromosomal looping
However, disrupting interactions between regulatory regions and promoters is not the only way non-coding variation can influence gene expression. Non-coding variation can also disrupt larger units of chromatin organization, known as topologically associated domains or TADs. A TAD is a stretch of DNA sequence (up to 1Mb) where regions inside the TAD are more likely to interact with each other than with regions outside of the TAD. In other words, there is a high degree of chromosomal looping within the confines of the TAD. If a variant disrupts the TAD boundary, then enhancers that normally only interacted with its wildtype target genes within the TAD would be open to influence the expression of genes outside the TAD (Figure 2). This is sometimes referred to as “enhancer hijacking.” A classic example of enhancer hijacking is polydactyly, a condition which results in more than five fingers or toes on the hand or foot. Polydactyly is caused by a deletion of a TAD boundary, which results in a cluster of enhancers associated with limb development interacting with genes that they would not interact with had the boundary been in place5. Depending on which TAD boundaries are deleted, the enhancers can interact with different genes leading to different phenotypes such as F-syndrome or Brachydactyly.
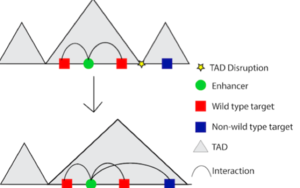
Figure 2: Disruption of TAD boundaries
New technologies and computational approaches are emerging for detecting chromosomal interactions genome-wide, leading to new and exciting opportunities to explore the role of regulatory variation in the context of the 3D genome. Moving forward, our attention should be focused on understanding the role of non-coding disease and trait-associated variants that have been identified in previous GWAS studies. We should use these technologies and computational approaches to map non-coding GWAS variants to their target genes. It is also important to investigate the effects of the non-coding variation on the target genes in the context of the disease or trait. Understanding the relationship between the 3D genome and regulatory variation will allow us to interpret much of the known GWAS variants by linking the variants directly to known and novel candidate genes for a wide-range of diseases and traits.