Genomics: Insight

An Overview of Sickle Cell Anemia
Pathophysiology, Symptoms, and Treatment
Symptoms and Pathophysiology
Sickle cell anemia (SS) is an autosomal recessive inherited red blood cell disorder that produces a wide variety of symptoms ranging from chronic hemolytic anemia, inflammation, vaso-occlusions, stroke, fatigue, and 2acute painful crisis (Platt 2008). Hemolytic anemia is a condition where blood cells in the body are destroyed at a faster rate than they are produced, making it difficult for the body to carry oxygen. Vaso-occlusions occur when the flow of blood is blocked from particular parts of the body. SS symptoms have a wide degree of pain variation based on the severity of the sufferer's disorder and environmental factors.
The symptoms of sickle cell anemia are exacerbated by dehydration, temperature extremes, vigorous exercise, illness/infection, and recreational drug use (Stuart and Nagel 2004). This genetic disease is highly prevalent, very painful, and has no known accessible cure. 1 in 700 newborns of African descent are shown to be affected by sickle cell anemia (Platt 2008). The reason this disease is highly prevalent is due to the advantages of having one copy of the sickle cell allele. This mutation is a 1balanced polymorphism in which one copy of this allele protects heterozygous carriers from 3Plasmodium falciparum malaria (Stuart and Nagel 2004). A balanced polymorphism occurs when there are heterozygous advantages to having one copy of each allele. Individuals with one copy of the sickle cell allele are shown to be protected against malaria due to an increase in a particular enzyme by cells with sickle cell hemoglobin. This enzyme causes the red blood cells to have an increased tolerance against malaria (Friedman 1978).
Sickle cell anemia is caused by two copies of the sickle hemoglobin allele (HBS) in the hemoglobin-beta gene on chromosome 11, resulting in a point mutation that alters the structure of hemoglobin, which is the protein that carries oxygen in blood cells and is made up of 2 pairs of polypeptides (Cole-Strauss et al. 1996). Normal hemoglobin heterotetramers contain 2 alpha chain polypeptides and 2 beta chain polypeptides. The sickle cell allele changes the normal beta hemoglobin polypeptide to a sickle hemoglobin polypeptide (HBS). This is harmful to red blood cells because at low oxygen concentrations sickle hemoglobin peptides form a linear polymer chain that becomes very rigid, and causes the sickle shape of the cell (Figure 1). This sickling is more common in small capillaries where there are low amounts of oxygen and it is reversible. However, over time the red blood cells can become stuck in the sickled shape from continuous sickling.
...at low oxygen concentrations sickle hemoglobin peptides form a linear polymer chain that becomes very rigid, and causes the sickle shape...
Sickled cells are unable to move through capillaries or bind to oxygen as easily as normal adult red blood cells. Sickled cells have a high affinity to bind to other cell membranes and venous endothelium causing a reduction of blood flow (Stuart and Nagel 2004). This stickiness causes the vaso-occlusions that the sufferer experiences (Figure 1). Vaso-occlusions can cause blockages in micro and major vessels throughout the body and in the brain which can lead to stroke (Platt 2008). The body will target and destroy the sickled cells, but they are produced at such a high rate that the sufferer will often experience hemolytic anemia. The symptoms of sickle cell anemia work together to form a feedback loop that increases the severity of the disease.
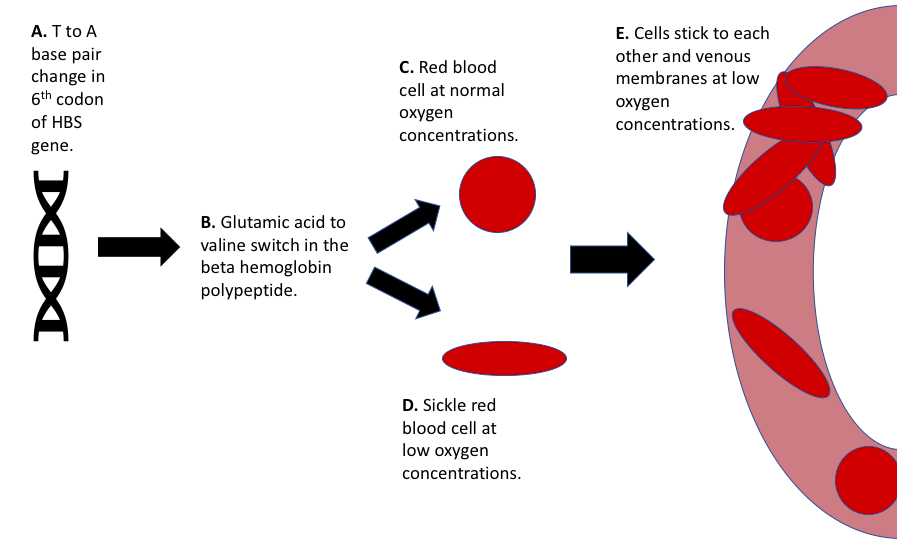 Figure 1: Pathophysiology of sickle cell anemia.
Figure 1: Pathophysiology of sickle cell anemia.This graphic shows the pathway of sickle cell anemia from gene to cell and its role in vaso-occlusion.
Treatment
Sickle cell anemia is a difficult disorder to treat since there are few physical signs of the disease other than the sufferer's intense pain. It is important that health care providers are well educated on the recognition of sickle cell anemia and disease management. There are few cures for sickle cell disease, but there are many treatment options. These options include blood transfusions, nutritional dieting, pain management, 4>ion channel blockers, 5nitric oxide, hydroxyurea, gene therapy, and 6hematopoietic cell transplantation (Stuart and Nagel 2004). The only known “cure” for sickle cell would be hematopoietic cell transplantation which is very costly, requires a close genetic match, and has a death rate of 5% (Stuart and Nagel 2004). This type of transplantation takes bone marrow/stem cells from a donor and transplants them into the patient's bloodstream. It is highly unlikely that a person affected with sickle cell disease will receive a hematopoietic cell transplantation due to the cost and rarity of finding a matching donor. The other treatment options can alleviate the symptoms of sickle cell anemia, but none will cure it. Gene therapy, such as LentiGlobin, is a viable option for afflicted persons in the future, however, it is not a widely available or cost effective approach presently (Figure 2).
LentiGlobin gene therapy works through the transplantation of hematopoietic cells that have been transduced with a lentivirus vector to produce antisickling hemoglobin through steric inhibition (Kanter et al. 2021). A lentivirus is an RNA virus that uses reverse transcriptase to transform its RNA to DNA and merge it with the host's DNA. This form of treatment directly modifies the patients DNA to produce anti-sickling hemoglobin. This hemoglobin has a similar oxygen affinity to normal adult hemoglobin and can significantly improve the symptoms of SS. One study found that the median concentration of hemoglobin increased from 8.5g/dl to 11g/dl or higher and seemed to take effect in 85% of red blood cells in the patient (Kanter et al. 2021). None of the patients treated with LentiGlobin had any vaso-occlusive events from 6-36 months post treatment (Kanter et al. 2021). Lentiglobin gene therapy is still in development and may be a viable treatment option for sickle cell anemia, however, treatment price is estimated to be over a million dollars per patient and long term effects of LentiGlobin have yet to be determined.
None of the patients treated with LentiGlobin had any vaso-occlusive events from 6-36 months post treatment.
As an alternative to the gene therapy treatment options, hydroxyurea treatment is very affordable and effective at combating the symptoms of sickle cell anemia. Hydroxyurea is shown to increase the amount of fetal hemoglobin in blood cells (Stuart and Nagel 2004). Fetal hemoglobin has a high affinity for oxygen and can be thought of as the opposite of sickle hemoglobin. This is due to hydroxyurea's cytotoxic effects on hemoglobin. Hydroxyurea has the ability to bind to ribonucleotide reductases' two iron molecules inactivating a tyrosyl radical (Platt 2008). This means that since sickle cells arise from rapidly dividing HBS blood cells, they are likely to be suppressed by hydroxyurea; while the slowly dividing fetal hemoglobin blood cells are expressed. Individuals with high amounts of fetal hemoglobin have milder symptoms of sickle cell anemia. This treatment is highly accessible and affordable when compared to other SS treatments and may aid in improving the quality of life for those affected with sickle cell anemia.
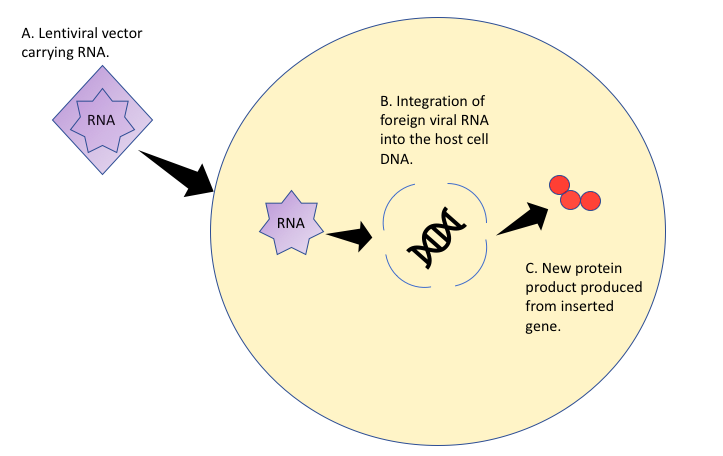 Figure 2: Mechanism of Gene Therapy Treatment with a Lentiviral Vector.
Figure 2: Mechanism of Gene Therapy Treatment with a Lentiviral Vector.Gene therapy using a lentivirus vector integrates particular genes into the patients DNA using viral RNA. A new protein product is produced from the inserted genes sequence.
Terms
- 1Balanced Polymorphism: When two different versions of a gene are stable in a population due to heterozygous advantages of having one copy of each allele.
- 2Acute Painful Crisis: Recurrent painful bouts that are usually caused by vaso-occlusions.
- 3Plasmodium falciparum Malaria: A deadly protozoan parasite that is transmitted into human blood via mosquito bite and causes malaria.
- 4Ion Channel Blockers: A molecule is used to prevent ion channels from opening; examples are lidocaine, prilocaine, etc.
- 5Nitric Oxide: A molecule that helps regulate vasodilation, meaning that it assists in widening blood vessels and increasing circulation.
- 6Hematopoietic Cell Transplantation: A bone marrow/stem cell transplant. This procedure puts healthy stem red blood cells into the patient's bloodstream.
References
- Cole-Strauss, A., et al. 1996. Correction of the Mutation Responsible for Sickle Cell Anemia by an RNA-DNA Oligonucleotide. Science. 273 (1386-1389). https://europepmc.org/article/MED/8703073
- Friedman, M. 1978. Erythrocytic Mechanism of Sickle Cell Resistance to Malaria. Proceedings of the National Academy of the United States of America. 75 (1994-1997). https://www.pnas.org/content/75/4/1994
- Platt, O. 2008. Hydroxyurea for the Treatment of Sickle Cell Anemia. The New England Journal of Medicine. 358 (1362-1369). https://www.nejm.org/doi/full/10.1056/NEJMct0708272
- Stuart, M., Nagel, L. 2004. Sickle-Cell Disease. The Lancet. 364 (1343-1360). https://pubmed.ncbi.nlm.nih.gov/15474138/
- Kanter, J., et al. 2021. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. New England Journal of Medicine. https://www.nejm.org/doi/full/10.1056/NEJMoa2117175
About the Author
Camryn Middlebrooks is a senior undergraduate student studying biology at the University of North Florida. Her interests range from conservation biology to genetics and epidemiology.